
A primer to Alphafold
Arguably the best protein structure prediction software in the market. Title inspired by @owlposting

Note: This post has been written more for a wet-lab scientist who has little prior knowledge about these tools, as opposed to a computational protein engineer.
Introduction
What is AlphaFold
Alphafold is an AI tool developed by Google DeepMind that can be used to predict protein 3D structures solely from their sequence. In other terms, all you input is the amino acid sequence of a protein, and you get a computational prediction of the structure of how the protein folds ( with confidence levels for each part of the protein), which you can then use for further downstream analysis, like docking, etc. The breakthrough: predicting 3D protein structure from sequence
note: all references to alphafold refer to alphafold-3 the latest version, unless specified otherwise.
How does Alphafold achieve this?
Here is a rudimentary explanation of what AlphaFold does under the hood to predict protein structures. (This part can be skipped if you only intend to understand how to use alphafold to the fullest)
Step 1: Input your amino acid sequence.
Step 2: AlphaFold searches sequence databases (UniProt, BFD, etc.) to find homologous proteins and builds a Multiple Sequence Alignment (MSA). This captures evolutionary constraints—if residues co-vary across species (e.g., both change from positive to negative together), they’re likely interacting in 3D space.
Step 3: Next, Alphafold searches the structure templates in places like databases like (PDB) to find proteins with known 3D structures similar to your target. These help to serve as structural ‘templates’ while generating the 3D structure for your target sequence.
Step 4: Armed with the MSA and the structural templates, the Neural Network iteratively predicts 3d coordinates for each atom until it finally converges on a final structure.
Q: But what if you find the exact protein in the PDB structure? Does it essentially reinvent the wheel?
A: Yes, hence its always a good idea to first check if the structure exists before spinning up alphafold.
Q: But what about for something like orphan proteins where there dont exist good homologues?
A: Yes, alphafold struggles to predict them effectively. In essence, this is one major limitation of Alphafold. It struggles to accurately predict structures of certain proteins, like:
Highly variable regions (CDR loops in antibodies)
De novo designed proteins
Capabilities

(From the Alphafold 3 paper.)
Alphafold can model several possibilities, not just single protein structures. Such as single-chain predictions and Joint structure of complexes including proteins, nucleic acids, small molecules, ions, and modified residues.
Note: Previously, you needed to use AlphaFold-Multimer for protein complexes and interactions between chains (e.g., antibody heavy/light chains, receptor-ligand complexes); however, now you can use AF3 for everything.
Differences between Alphafold-2 and Alphafold-3
Alphafold 2 consists of a Novel Neural network architecture (Evoformer), which it uses to process MSA and pair representations, which it feeds to the structure modules, which directly output 3d coordinates for each atom

The alphafold 2 Network architecture
Alphafold 3, alongside the initial Evoformer model for processing of sequence alignment and templates, uses diffusion modules for final structure generation. This essentially means it starts from random atomic coordinates and iteratively “denoises” them into the final structure. We will explore its implications in a bit.
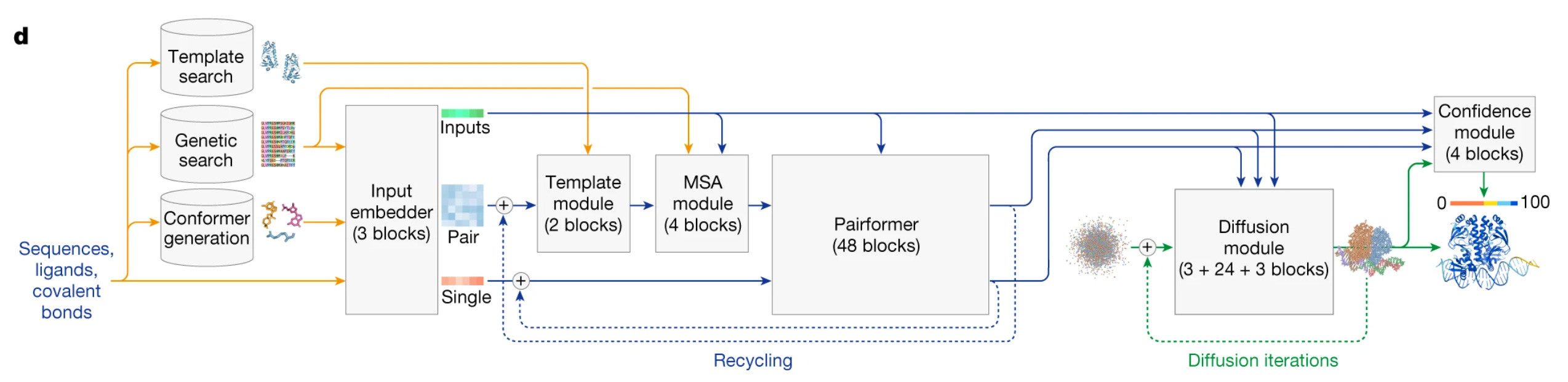
(Alphafold 3 architecture)
Limitations
However, like all AI models, there is no free lunch. One of them is that the model doesn’t respect chirality(a 4.4 violation rate, to be precise). At times, there are also cases of it producing clashing atoms in its structure predictions. These are mostly seen when predicting protein-nucleic complexes, for compounds having greater than 100 nucleotides and greater than 2,000 residues in total.
With the move towards a diffusion generative block for outputting 3D coordinates from AF2, we also see a change in the model, which outputs more hallucinations in disordered regions. With Alphafold-2, these low confidence regions had a trademark ‘ribbon-like’ structure; however, AF3 seems to instead completely hallucinate structures on its own at times.
It is also important to note that Alphafold has been trained on static structures as seen in PDB files, and hence does not generalize well to predicting dynamic conformational states as seen in solutions. To tackle this problem, the developers recommend generating a large number of predictions with different seeds and ranking them, particularly for antibody-antigen complexes.
Alphafold for the motivated user
For most people, the simple act of plugging in amino acid sequence(s) and downloading the PDB files and analyzing them in some visualization software like PyMol should suffice, which is pretty easy to do, hence I won’t go into that in here. This part should serve as a guide for people trying to do more funky things with Alphafold.
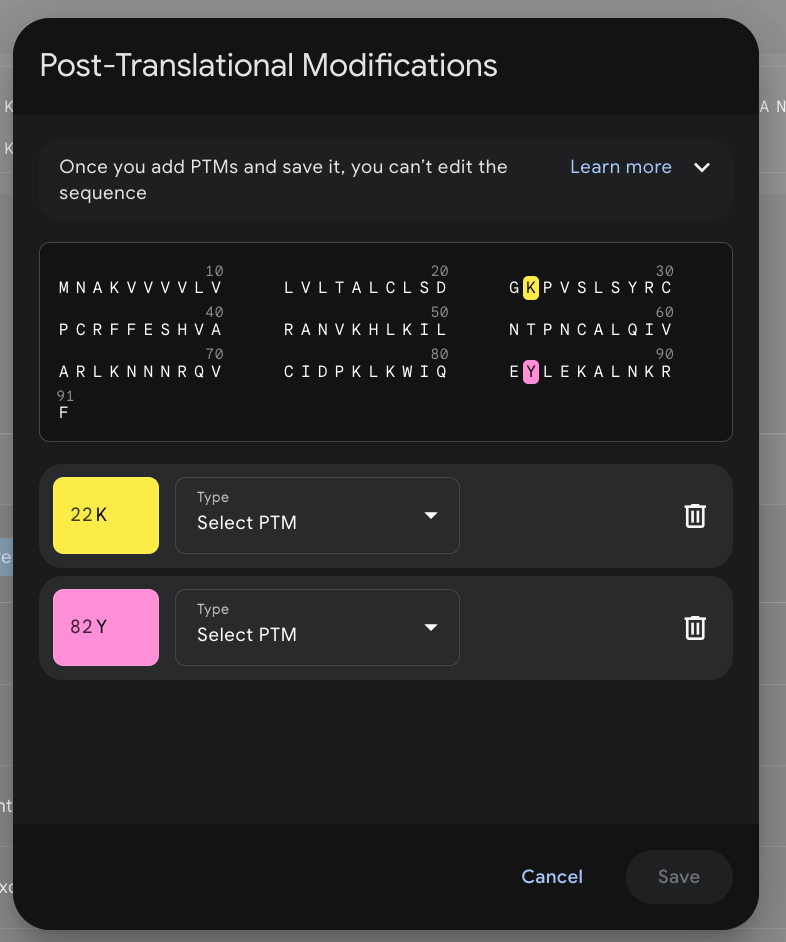
Post-Translational Modifications

(you can find them while clicking the three dots on the right side)
With Alphafold 3, you can specify the post-translational modifications for certain amino acids. This helps with understanding how certain PTMs (like glycosylation, for instance) can affect the proteins’ 3d conformations and show potential clashes or steric hindrances. There are certain tools like Phosphosite, which is a database for phosphorylation, acetylation, ubiquitination, etc, or something like NetPhos when experimental data is unavailable.
Template Settings
Alphafold allows you to control which PDB structures are used as templates. You have 3 options to choose from
Use PDB Templates up to a custom date: Allows you to specify exactly which structures AlphaFold can “see”.
Use PDB templates with default cut-off date (29/09/2021): This is the date AlphaFold 2 was trained on and prevents data leakage.
Turn off templates: Force AlphaFold to predict entirely from MSA evolutionary information.
Ideally, if you’re trying to validate your proteins, it’s a good idea to set a custom date for the PDB templates lest Alphafold use your own structure as a template. For truly de novo antibody design, for instance, it may be a good idea to completely turn off templates, as Alphafold may try to shift your structure towards more canonical structures.
Metric scores— all you need to know
PlDDT
Alongside the predicted structure, Alphafold also gives you a score for each part of the predicted structure, which helps you figure out which parts you can trust and which likely require further work.
PlDDT stands for Predicted Local Distance Difference Test. It measures the model’s confidence about the ‘local structure’ of the protein. A good rule of thumb is to go off the color scheme in Alphafold: blue high confidence, yellow = slightly disordered, and orange is = pretty disordered. While it’s a useful metric, it often falls short in the parts that really matte,r like the CDRs of antibodies or linker regions.
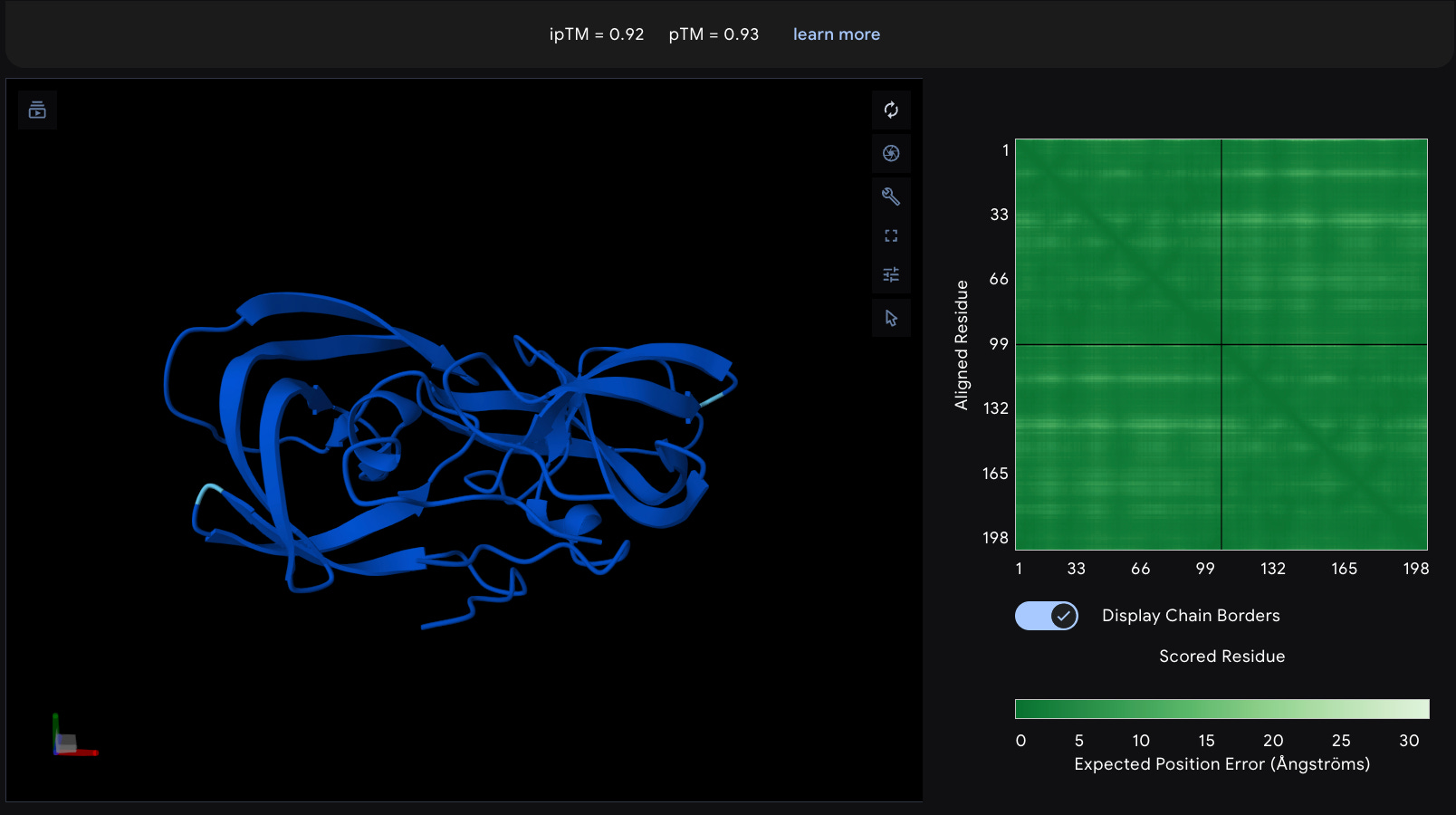
PAE
While pLDDT tells you about local confidence for each residue, PAE tells you about the relative positioning between different parts of your protein. The scale is typically 0-30 Ångströms. Ideally you see
Strong dark blue blocks along the diagonal (well-structured domains
Dark blue off-diagonal blocks connecting domains (domains positioned correctly and
For multimers: dark blue blocks between different chains (confidence in the interface)
(HIV Protease Homodimer, one of the most confident I’ve ever seen Alphafold)
Limitations:
However it is imperative to note Alphafold structures should be taken with a large grain of salt especially for GPCR’s. Here’s an example

(β2-Adrenergic Receptor + Gαs complex)
Notice how the β2-adrenergic receptor complexed with its G-protein shows dramatically lower confidence scores (ipTM=0.14) compared to the HIV protease dimer (ipTM=0.92). And we can see in the plot, while AlphaFold can predict the G-protein structure reasonably well, the interface between the receptor and G-protein (off-diagonal regions) is almost completely white.
Feel free to add your questions in the comments, and I’ll try my best to answer them or make a follow-up post!
That 'solely from their sequence' part is amazing so clear!
do you think learning structures might actually be detrimental to solving the orphan problems? seems like they would bias the model towards solutions that would be homologous and therefore incorrect. is rl or some other learning method a better approach?